
Status: Fund open
The LifeArc Philanthropic Fund
The Fund provides grants and funding to academic researchers working to advance new treatments and diagnostics for rare diseases.
As an independent, self-funded charity, we have been bridging the gaps in biomedical innovation for 25 years. We want to accelerate scientific breakthroughs that could deliver new interventions for patients with rare diseases. That is why we formed the LifeArc Philanthropic Fund in 2017.

Overview
Around 7,000 rare diseases affect about one in 20 people worldwide. That includes 3.5 million people in the UK. These conditions are often chronic, life-threatening and isolating for the sufferers and their families.
Only around 400 of these indications have licensed treatments and that number is only increasing slowly, as industry is reluctant to invest without a likely financial return on investment. In addition, some ultra-rare diseases however may never yield any commercial return, as patient numbers are too small.
Funds for research on rare diseases are scarce, particularly for therapeutic development, as industry is reluctant to invest without a likely financial return on investment.
LifeArc’s Philanthropic Fund awards grants to academics who have promising projects focused on research into therapeutics, devices or diagnostics that could support people who are living with a rare disease. We have a history of supporting the delivery of transformational therapeutics and are always looking for new ways to progress promising science into therapeutics and diagnostics. The research we fund must also have a credible path to patients.
Where possible, we welcome the opportunity to partner with a charity, patient-group or industry partner who shares our aim of addressing the need for solutions to rare disease.
Our funding allows research projects to remain longer in academia and move further along a development pathway – by which point they may be more attractive to follow-on investors.
Apply
If you have a project focused on research into therapeutics, diagnostics or devices that could support people living with a rare disease, please get in touch.
The story so far
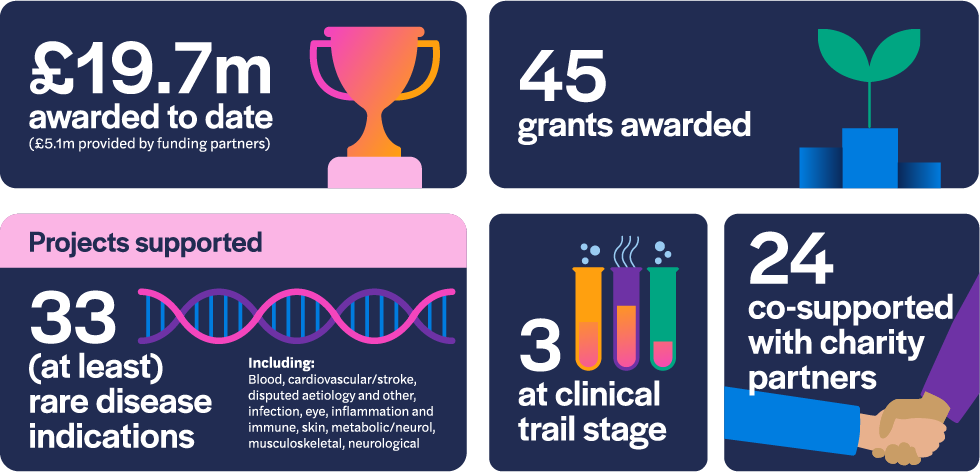
As of December 2022, the LifeArc Philanthropic Fund has awarded £14.6 million to 45 research projects since 2017. 24 of those grants have been to projects co-supported by our charity partners and three of the projects are at the clinical trial stage of development. The awarded grants comprise research that addresses 33 rare disease indications.
Funding to progress promising science
We are always looking for new ways to progress promising science. We evaluate promising approaches that are made by academics and researchers in the United Kingdom. Projects should be underpinned with a strong, scientific rationale in addressing a rare disease medical need and must be target driven, with a credible delivery plan and led by an academic.
LifeArc does not require a share of intellectual property (IP) or revenue return. But we need to know that, if the work is successful, you or your partners have the IP rights to commercialise the product so it can reach the patient.
We will help advance your innovation, supporting you and your research team as you strive to turn science into new therapeutics and diagnostics.
Application process
We review all submissions of non-confidential information and, if there is a good fit with our criteria, we will arrange a meeting with the project lead to learn more about the work.
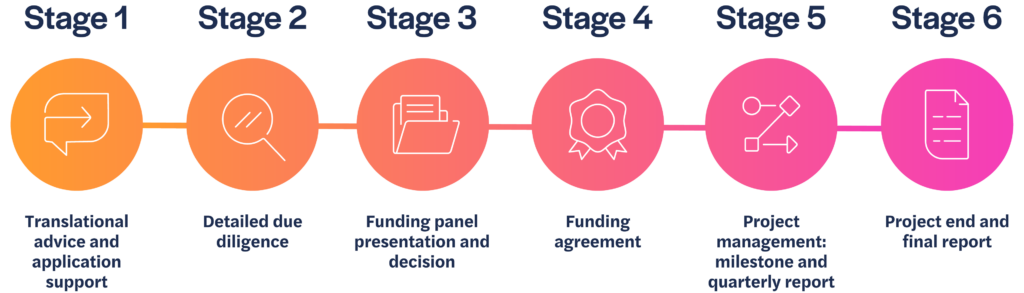
Stage 1
If we agree to move forward, we will ask you to complete a more detailed application. Our Philanthropic Fund team will support applicants going through this phase of the process, to ensure the proposal is complete and presented effectively.
Stage 2
Once submitted, we will conduct due diligence on the information provided, and gain external peer review, sharing comments back with you to address if you wish.
Stage 3
An application package consisting of your application and all related peer review information is considered by the funding panel. Funding decisions are made by the panel twice a year, usually in December and June.
Stage 4
If the panel agrees to fund your project, we will formalise the funding agreement and project milestones.
Stage 5
We will continue to work with you to support and manage your project so you achieve your agreed milestones on which the funding depends.
Stage 6
We will review the final results of your project and discuss if, and how, this research could be taken forward.
Panel members
- Robin Ali PhD FMedSci (Chair)
- Professor Mimoun Azzouz
- Kev Dhaliwal
- Stuart Forbes
- Volker Germaschewski
- Professor Manju Kurian
- Dr David Matthews
- Rose Sheridan
Funded projects
Get inspired by some of the research already funded
As of December 2022, the LifeArc Philanthropic Fund has awarded £14.6 million to 45 research projects since 2017. Click on the items below to find out about the projects we have funded and which are making progress towards translating science into treatments or diagnostics for the following rare and ultra-rare diseases:
Amyotrophic lateral sclerosis and frontotemporal dementia
| Information | Answer |
|---|---|
| Project title: | SRSF1-targeted gene therapy for C9ORF72-linked Amyotrophic Lateral Sclerosis and Frontotemporal Dementia |
| Principal investigator: | Dr Guillaume Hautbergue, University of Sheffield |
| Co-investigators: | Prof Mimoun Azzouz and Prof Dame Pamela Shaw, University of Sheffield |
| Start date: | 18 September 2020 |
| Duration: | 36 months |
| Amount funded: | £513,141 |
| Partner funding: | £50,000 co-funding contribution from the MND Association |
Amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD) are incurable and fatal adult-onset neurodegenerative diseases. Someone in the world is diagnosed with ALS or FTD every few minutes.
People with ALS lose nerve cells in the motor system – leading to progressive paralysis and death, usually within 2-5 years of the first symptoms.
In people with FTD, nerve cells in the frontal and temporal lobes of the brain die, affecting cognitive function and behaviour. A fault in a gene known as C9ORF72 is the most commonly known genetic cause of both conditions.
There is currently no treatment for FTD. The current treatment for ALS patients, riluzole, only marginally extends survival – typically by around three months – and does not relieve the symptoms of progressive paralysis.
In both conditions, the gene is copied into the cells by a protein, which the team identified as SRSF1.
In C9orf72-linked ALS and FTD, one the main drivers of disease is the transport of pathological, C9orf72 repeated RNA molecules from the nucleus to the cytoplasm where neurotoxic dipeptide repeat proteins (DPRs) are generated. The team identified a particular protein called SRSF1 which binds to the pathological repeated RNA molecules and transports them out of the cell centre, effectively overriding the gatekeeping machinery within the nucleus by opening a back door.
The team had already shown that, by using a gene therapy comprising interfering virus vehicles to reduce the levels of SRSF1 protein, there was a reduction in nuclear transport of pathological RNA molecules and generation of neurotoxic DPRs – addressing a root cause of the diseases.
In the laboratory, this gene therapy approach promotes the survival of patient-derived nerve cells and has no detrimental effects at the genome-wide level or in control cell cultures.
The team also demonstrated that SRSF1 depletion prevents paralysis in a fly model of C9ORF72-ALS/FTD, and also that this intervention is safe in healthy mice.
The purpose of this project is to conduct further studies to optimise the gene therapy treatment strategy ahead of developing a clinical trial application as follows:
- Evaluate the best delivery route for gene therapy.
- Define the dose of gene therapy needed to achieve the best reduction of SRSF1 in a mouse model of C9ORF72 disease.
- Conduct a pilot study to assess the therapeutic efficacy and any potential long-term adverse effects of the therapeutic gene therapy.
- Identify the biological processes which are modulated in the mouse brain after treatment and compare them to data available in patient-derived brain cells.
- Seek advice from regulatory bodies for a pre-clinical package to facilitate future clinical development.
The Motor Neurone Disease Association (MND Association) contributed £50,000 to the project.
This new research will be conducted in collaboration with the Cell and Gene Therapy Catapult which will provide support with designing the clinical vector and a detailed non-clinical safety strategy as well as regulatory advice to guide the team at the University of Sheffield through interactions with the Medicines and Health Regulatory Agency (MHRA).
Congenital factor VII deficiency
| Information | Answer |
|---|---|
| Project title: | Gene Therapy for Congenital Factor VII Deficiency; Final IMPd/IND Enabling Studies |
| Principal investigator: | Amit C. Nathwani, UCL Cancer Institute, Department of Haematology |
| Co-investigators: | Prof Flora Peyvandi; Fondazione IRCCS Ca’ Granda, Italy. Dr Kaan Kavakli; EGE University Childrens Hospital, Turkey. Prof Johannes Oldenberg; University Clinic Bonn, Germany. Dr Mary Mathias; Royal Free Hospital & GOSH, UK. Prof Edward Tuddenham; UCL. Prof Guglielmo Mariani; Westminster University, UK. Prof Amy Shapiro; Indiana Hemophilia & Thrombosis Center, USA. Dr Ulrike Reiss; St Jude Children’s Research Hospital, Memphis, USA. Dr Andrew Davidoff; St Jude Children’s Research Hospital, Memphis, USA. |
| Start date: | 20 March 2019 |
| Duration: | 21 months |
| Amount funded: | £811,582 |
| Partner funding: | $2M from Hemophilia of Georgia to St Jude Children’s Research Hospital, Memphis, USA |
This study aims to move forward the development of a cutting-edge gene therapy approach for severe factor VII deficiency (SF7D), a life-threatening bleeding condition characterised by a deficiency of clotting factor VII – a protein essential for the blood to clot normally. The condition is linked to a recessive chromosome and is thought to affect around 1 in 500,000 people.
The liver is one of the prime targets for gene therapy to correct defects in a variety of clotting factor genes, but gene therapy for this condition has not been explored due to its rarity and the perceived difficulty in intervening early enough in life to prevent permanent organ damage and death.
This project is using the same virus vector platform technology that supported successful gene therapy in people with a related condition, haemophilia B, by mediating the continuous expression of a transgenic protein in these patients.
The team has developed a more potent form of the virus vector containing a codon optimised human FVII cDNA under the control of a small liver-specific promoter. The new virus vector has shown to greatly increase the transgene expression and preclinical data has enabled the potential therapy to be granted an orphan drug designation (ODD).
This study proposes/will undertake phase I/II clinical trials of the orphan drug in adults, potentially followed by trials in adolescents and children.
The team has established a wide-reaching clinical research network which, together with swift trial design and access to clusters of patients, improves the prospect of successful gene therapy for the condition.
Dravet syndrome
| Information | Answer |
|---|---|
| Project title: | AAV9-mediated gene targeting of natural antisense transcript as a novel treatment for Dravet Syndrome |
| Principal investigator: | Dr Rajvinder Karda, University College London (UCL) Institute for Women’s Health |
| Co-investigators: | Simon N Waddington, UCL. Stephanie Schorge, UCL. Helen Cross, UCL. |
| Start date: | 1 May 2020 |
| Duration: | 36 months |
| Amount funded: | £575,831 |
Dravet syndrome is a rare neurological condition that is often described as a complex form of epilepsy.
Children and adults with this lifelong condition will experience severe seizures that are difficult to control. Individuals with the condition may also suffer from varying degrees of intellectual disability and a spectrum of associated health conditions (known as ‘comorbidities’) – which may include autism, ADHD, challenging behaviours and difficulties affecting their speech, mobility, eating and sleep.
80% of patients with the condition carry a mutation in the SCN1A gene which encodes a sodium ion channel, NaV1.1, which functions to maintain normal firing patterns in neurons. Mutated NaV1.1 channels in Dravet syndrome patients do not function properly, which results in abnormal neuronal activity.
There is no cure and current treatments have limited benefit, as well as side-effects.
With this funding, Dr Karda aims to develop a gene therapy for Dravet syndrome. The approach involves delivering a gene element, using a viral vector, in order to increase healthy SCN1A expression and thereby restore the number of functional sodium ion channels, which in turn should normalise neuronal excitability.
The project will test the novel gene therapy in mice which have a deletion in the Scn1a gene (mouse equivalent gene) and therefore are a model of Dravet Syndrome. The Dravet mice, like the human Dravet patients, contain one functional gene and one non-functional gene and show seizures, cognitive impairment and premature death, which makes it a clinically relevant model of disease. Treatment is expected to upregulate the Scn1a gene, producing more functional sodium ion channels, which should restore normal neuronal activity.
The aim is to generate an optimised, clinically relevant gene therapy treatment which has demonstrated robust efficacy and safety in a clinically translatable model of the Dravet syndrome disease.
The gene therapy has the potential to treat all patients with an SCN1A mutation that causes loss of function, regardless of the specific mutation. It could potentially reduce or completely stop seizures, cognitive impairment, ataxia (an inability to coordinate muscle movements) and premature death.
Idiopathic intracranial hypertension
| Information | Answer |
|---|---|
| Project title: | Non-invasive MRI of Blood-Cerebrospinal Fluid Barrier Function: A Breakthrough Translational Method to Improve Treatment for Idiopathic Intracranial Hypertension |
| Principal investigator: | Dr Jack Wells, University College London |
| Co-investigators: | Prof Alexander Gourine, University College London |
| Start date: | 18 September 2020 |
| Duration: | 24 months |
| Amount funded: | £146,781 |
Idiopathic intracranial hypertension (IIH) is a rare disease that most commonly affects women of childbearing age. Patients experience extremely painful headaches and can suffer loss of vision that, for many, leads to permanent damage to their eyesight.
The condition causes fluid pressure to build up in the brain. The primary source of this fluid pressure is a structure in the brain called the choroid plexus. While drugs can control the function of the choroid plexus, research has revealed that the benefits of these drugs are highly unpredictable.
This reflects a lack of data measuring how effectively the drugs affect the function of the choroid plexus and the resulting change to brain fluid pressure.
The team’s laboratory had previously invented the first non-invasive technique, using MRI, to measure the function of the choroid plexus – the part of the brain that these drugs seek to target.
In this study, the team is using the new, non-invasive technique in combination with intra-cranial pressure monitoring techniques to see how a range of drugs already approved for IIH affect choroid plexus function and brain fluid pressure in anaesthetised mice. This will allow the team to define how drugs affect choroid plexus function and brain fluid pressure for the first time.
The resulting data will fill a gap in knowledge about the mechanisms that underlie drug treatment strategies for IIH and provide a strong foundation for improving drug treatment for patients.
The technique has potential as an enabling technology and further development through this project could enable better drug assessment in other indications linked with raised ICP and ChP dysfunction.
View the fund award conditions.
FAQs
What are the criteria for rare disease funding?
What is the LifeArc Philanthropic Fund looking for?
For applications to be funded they must:
- address a rare disease medical need
- have a strong scientific rationale
- be target-driven projects with milestones and a credible delivery plan
- have a route to patient
- have, or will have, Intellectual Property to secure the route to patient
How can LifeArc help me take my project forward?
If you have a discovery, technology (e.g. prototype device, potential therapeutics, novel drug targets) or materials that you think might have a commercial application, please contact us with non-confidential information about your project. If successful, you can access the expertise, facilities and resources needed to develop your research in a commercial environment.
Do you provide financial support for drug screening?
LifeArc can provide funding and support for academics as well as for early-stage spin out companies with strong intellectual property positions and a market-driven proposition. Please contact us and provide some non-confidential details about your project.
Can you help fund early phase clinical trials?
We may be able to help you if you are an academic institution or nascent spin-out. Please get in touch.
I would like to conduct research into a surgical procedure. Can LifeArc provide funding?
We are not able to support research into various disciplines of surgery. We refer you to the Surgery Research Society (SRS) for sources of funding.
Can LifeArc fund my PhD?
LifeArc does not directly fund students. However, through our collaboration with the Research Council UK, we provide support for some PhDs in disease areas relevant to our charitable objectives. When available, these programmes are usually advertised on the relevant academic notice boards, university websites and on online PhD databases.
Can LifeArc fund my academic research project?
We award grants to academic institutions, either directly from LifeArc or in combination with funds from one of our charity partners.
How do you assess projects for funding?
Academics need to submit a non-confidential proposal for initial review. A funding panel, made up of translational scientists and clinicians from academia and from industry evaluate, projects for funding twice a year. Find out more about the application process.
Follow us us on LinkedIn or Twitter for updates on new and repeat funding calls.
Can you help me get onto a medical trial?
Unfortunately, we are not in a position to recruit people to clinical trials. Please ask your doctor or a patient organisation if they know of any clinical trials that you may be eligible to join.
How do I license your technology?
We have a wide range of life science licensing opportunities and technologies available. We are also responsible for the technology transfer of patented and protected technologies from the MRC and other academic and charitable organisations. Find out about the licenses available or find a contact within our Technology Transfer team to talk to.
What is the assessment process?
Decisions on which applications will be funded will be reached via a competitive process comprising an expert scientific panel made up of translational scientists and clinicians from academia and from industry who will, in turn, be supported by peer review. As the Fund-supported projects progress LifeArc will continue to support the research group.
Can I reapply if unsuccessful?
Feedback is provided with all funding decisions. In the event that the funding panel would welcome the resubmission, this will be outlined.
Can I apply with a co-funder?
Yes, co-funded applications are welcome with the appropriate collaboration terms outlined. Funding support will be awarded to the academic institution.
Does LifeArc expect a revenue share?
LifeArc will not ask for any financial return or intellectual property from projects supported by the Philanthropic Fund, allowing researchers to focus purely on delivering a result that benefits patients.
What are the reporting requirements?
Awardees are required to provide an update report on a quarterly basis from the project commencement date.
What happens in the event of delays in progressing and completing the project?
The Philanthropic Fund team works closely with funded projects throughout the project term to ensure the project commences as planned and all milestone objectives are met. The Philanthropic Team will work with the awardee to address any issues.
Contact the Philanthropic Fund team
In submitting your personal data via this form, you consent to being contacted via the details provided so that your enquiry can be responded to. If you would like your data to be removed, please email dataprivacy@lifearc.org.
Please see our Privacy Policy in relation to the personal data you submit to us through this page.
Latest news
-

LifeArc launches £40m research centres that will unlock new tests, treatments and cures for people living with rare diseases
Read more: LifeArc launches £40m research centres that will unlock new tests, treatments and cures for people living with rare diseases -

First-of-a-kind plan announced to get more children access to cutting-edge, proven gene therapy treatments for rare diseases
Read more: First-of-a-kind plan announced to get more children access to cutting-edge, proven gene therapy treatments for rare diseases -

New £6.2m partnership will help to eliminate a deadly disease affecting children and vulnerable people in Kenya
Read more: New £6.2m partnership will help to eliminate a deadly disease affecting children and vulnerable people in Kenya